
Chassis selection is a critical early decision in any synthetic biology project, particularly when working in environments beyond Earth. As we push the boundaries of In Situ Resource Utilization (ISRU), the need to choose organisms that can efficiently harness local minerals and resources becomes paramount. Traditional laboratory organisms like E. coli dominate research due to their well-understood biology. However, their suitability for non-terrestrial environments often requires extensive genetic engineering to meet the demands of space-based resource constraints.
To address this challenge, our team developed Astrolabe, a software tool designed to assist researchers in selecting the most appropriate chassis organism based on specific environmental resources and physiological conditions. By leveraging available resources and minimizing the engineering required, Astrolabe provides a ranked list of suitable organisms, offering a streamlined path to achieving desired biological outputs with maximum efficiency.
Named after the ancient instrument that guided explorers across the seas, Astrolabe is set to guide the next generation of explorers in space. Just as the medieval astrolabe was a tool for navigation and discovery, our modern-day Astrolabe will aid scientists and engineers in navigating the complex landscape of biological resource utilization in space.

Find the software hosted here: http://astrolabeiitm.duckdns.org/
Inspiration
During the early stages of ideation for our project, one of the most significant challenges we encountered was selecting suitable host organisms, particularly those capable of solubilizing silicates and utilizing them in extraterrestrial environments.
This task was far from trivial, as identifying organisms with the ability to interact with and metabolize silicate-based minerals required an exhaustive review of both microbiology literature and resource availability data. After conducting an extensive literature review, we eventually identified a handful of organisms that could meet our specific use case. However, the process was both time-consuming and difficult, underscoring a broader issue within the field.
In our search for a more efficient way to select organisms that could maximize resource utilization, we assumed that a software tool already existed to recommend chassis organisms based on given environmental resources. To our surprise, no such tool was available. This gap in existing SynBio tools sparked the idea to create a solution ourselves.
We embarked on developing Astrolabe, a software designed to ease this burden by providing a ranked list of organisms tailored to specific environmental conditions and resource constraints, streamlining chassis selection and significantly reducing the time required for manual literature reviews and organism screening.
Pipeline
Overview
Our software pipeline facilitates analyzing and identifying synbio hosts based on available resources and growth conditions as user inputs. The aim is to gather relevant resources from the user, perform pre-processing, search for metabolic data, find resource utilization pathways, and ultimately provide a ranked list of potential chassis that comprise these pathways.
User Input and Preprocessing: The process starts with the user entering the common name of a molecule and selecting its corresponding ChEBI ID from a list. We’ve chosen the ChEBI standard for input as it provides a cataloging of a wide array of chemical formats and offers easy code integration via its API. The chosen ID can then be added to the Resources list. Also, the user can input query-related information, such as required physiological conditions (e.g., temperature, pH)
Metabolite Search and Matching: Executing a search for specific reactions of these resource compounds and searching for metabolic pathways comprising these reactions in several databases, such as BioCyc, UniProt (Universal Protein Resource), KEGG (Kyoto Encyclopedia of Genes and Genomes) and BRENDA (Braunschweig Enzyme Database). These metabolic pathways are then matched to potential hosts
Scoring Function: The identified chassis are evaluated and ranked based on two key criteria: resourcefulness and survivability. Resourcefulness measures the utility of the organism as a chassis, determined by the number of pathways identified within it. Survivability assesses how well the organism's optimal growth conditions, such as temperature and oxygen requirements, align with the user's input. The physiological information is obtained from BacDive and MediaDive. The closer the match, the higher the ranking for survivability.
Output: Using the scoring function, a ranked list of organisms that can be engineered for In Situ Resource Utilization (ISRU) is generated. The top organisms are selected based on their resource utilization pathways and adaptability to various conditions. The associated pathways and potential use cases are presented.

BioCyc
Overview of BioCyc
BioCyc is a collection of Pathway/Genome Databases (PGDBs) for model eukaryotes and for thousands of microbes, plus software tools for exploring them. It contains curated data from 146,000 publications. The reason BioCyc was picked as one of the databases was due to the unique pathways and metabolites in its catalog, including some inorganics and pathways involved in critical biogeochemical cycles, absent in other databases.
Accessing BioCyc Using Python's Requests Package
Python’s Requests package allows users to query web pages and collect the information on the page. First, we created a requests session, which allowed us to log in to BioCyc. This was necessary because BioCyc requires the user to log in before being able to access the database. After doing this, we queried various payloads corresponding to each step of the workflow.
Mapping ChEBI IDs to Pathways and Organism Filtering
User-provided ChEBI IDs are converted to BioCyc IDs for compatibility with metabolic databases. The software then retrieves biochemical reactions involving the specified compounds, identifies associated metabolic pathways, and collects organisms documented to contain these pathways. To ensure relevance for synthetic biology applications, multicellular organisms under the taxonomic groups 'Metazoa' and 'Embryophyta' are filtered out, leaving only unicellular candidates. The remaining organisms are then passed through a scoring function, which ranks them based on their suitability as chassis organisms, optimizing for resource utilization efficiency and genetic tractability.

UniProt
Overview of UniProt
UniProt is a comprehensive protein sequence and functional information database. It provides detailed annotations for proteins, including their functions, structures, and interactions. UniProt is a crucial resource for researchers in fields such as genomics, proteomics, and bioinformatics. It hosts a wealth of curated data from various sources, allowing users to explore relationships between proteins, their corresponding organisms, and biochemical pathways. The database is especially beneficial for studies involving synthetic biology and metabolic engineering due to its extensive protein functional information.
Accessing UniProt Using Python's Requests Package
The Python Requests package enables users to easily query the UniProt REST API to retrieve protein-related information. The querying process begins by constructing a request URL that incorporates specific filters such as taxonomy and ChEBI identifiers. This step ensures that only relevant protein data is fetched from UniProt. The code provided utilizes a session with the UniProt API to access necessary protein data based on the given ChEBI IDs.
Mapping ChEBI IDs to UniProt Data
The process of querying UniProt involves the following steps:
-
ChEBI ID Input: Users input ChEBI IDs as strings, which are then processed to extract the relevant part for querying.
-
UniProt Query Construction: A query URL is constructed that targets specific organisms (e.g., bacteria) and filters for proteins that have been reviewed and are associated with the specified ChEBI ID as a cofactor.
-
Data Retrieval: The constructed URL is used to send a GET request to the UniProt API. The resource list in ChEBI ID format is passed via the request to get the organism with the corresponding number of pathways containing the resource of interest in it. If the request is successful, the response data is returned in JSON format.
Filtering Organism Data
Once the data is retrieved, it undergoes processing to filter out duplicates and irrelevant organisms:
-
Duplicate Filtering: The filter_duplicates function counts occurrences of each organism based on its scientific name, ensuring that each unique organism is represented only once in the results.
-
Relevance Filtering: The code further filters results based on the annotation score (indicating the likely accuracy of the data), retaining only those entries with a score greater than or equal to 3, and excluding any organisms classified as viruses.
Summary of Results
The final output is a counter of unique organisms that meet the specified criteria, along with their respective counts. The results provide insights into the distribution of proteins associated with the input ChEBI IDs, enabling researchers to identify the most relevant organisms for further study.
KEGG and BRENDA
Introduction
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a collection of databases dealing with genomes, biological pathways, and chemical substances. BRENDA (BRaunschweig ENzyme DAtabase) is the world's most comprehensive online database for functional, biochemical and molecular biological data on enzymes, metabolites and metabolic pathways. In the current architecture of the software, KEGG serves as the backbone for retrieving organismal data while BRENDA supplements enzyme data for deeper insights into metabolic pathways. KEGG and BRENDA are queried to retrieve enzymes and genes that act on the compound. Then, KEGG is queried to collect enzyme and gene information to identify organisms.
KEGG structures its database as a collection of interconnected sub-databases (KEGG Compounds, KEGG Enzymes, KEGG Genes, KEGG Organisms, and KEGG BRITE). Navigating through these sub-databases was unintuitive due to the lack of cohesive documentation explaining how the sub-databases cross-reference each other. After some iterations, we settled on a compound → enzyme → gene → organism approach, which proved more efficient. Following this, we have implemented a filtering step to include only those taxa that are relevant to synthetic biology, including Ascomycetes, Basidiomycetes, Diatoms, Euglenozoa, Green algae, Red algae, along with bacteria.
Accessing and Sorting Data
The KEGG subroutine required optimisations to overcome 2 major bottlenecks, namely query limits and the complexity of mapping enzymes to genes and genes to organisms:
-
Batching Queries: By sending requests in batches instead of individually, we reduced redundant queries and lowered overall processing time.
-
Multiprocessing: Multiprocessing is a computer paradigm in which two or more processors in a computer are simultaneously executing different portions of the same program. This technique allows for better utilization of available hardware and improves speed. We implemented Python’s multiprocessing library to run multiple independent queries in parallel. This was particularly effective since each query was isolated and could be processed independently. This optimization alone shaved around 30 seconds off the initial 4 minutes.
-
Local Searching: We discovered that KEGG uses a prefix in gene codes corresponding to organism codes. By downloading a local list of organism codes from KEGG, We were able to bypass querying the server for each gene, dramatically reducing the time spent on gene-to-organism mapping by eliminating network delays.
As not all compounds were documented in KEGG, we decided to complement the KEGG results by running the same compound queries through BRENDA, to see if additional enzymes could be identified. BRENDA uses the SOAP API architecture. SOAP is fundamentally different from the REST architecture used in KEGG, requiring an intermediary service provider to process requests, which added complexity. Due to improperly configured API endpoints and function prototypes, we were unable to sort the API calls in time. Thus, we resorted to using BRENDA’s CSV download functionality. We have constructed dynamic URLs to automate the download process of the EC enzyme information. Integration of BRENDA let us generate more comprehensive enzyme lists.
Graphical Overview

Scoring Function
Parameters
The scoring function takes the output from the different databases executing Metabolic Search, integrates and sorts the output to give the user the benefit of the different information sources.
The scoring function plays a crucial role in ensuring the user gets maximum utility from our software. We have identified the following factors to characterize and quantify the suitability of a potential chassis organism for the expressed use case.
Resourcefulness Score
This parameter quantifies the competence of the organism at utilizing the most number of metabolites of interest. The score is the sum total number of pathways/reactions/enzymes in an organism that contain the resources(s) of interest from all the 4 databases divided by the maximum number of pathways identified in a single organism across the databases amongst all the organisms. This normalizes the score and makes it simpler to scale using the weights. This information is supplied from the Uniprot, BioCyc, KEGG and BRENDA databases.
$$ \text{Resourcefulness score} = \frac{\sum_{database}^{n=4} \text{no. of pathways/reactions/enzymes}}{\text{maximum no. of pathways}}$$
Survivability Score
This parameter quantifies the suitability of the organism to grow in the environment desired by the user. Although microorganisms on Earth cannot bear extraterrestrial conditions, considerations like this can help increase the feasibility of the proposed project by, for example, reducing energy expenditure in maintaining the required temperatures in a bioreactor. We have considered the overlap in Temperature and pH ranges between the user input and the organism’s preferred ranges (normalized by dividing with respective input ranges), supplied from the BacDive and MediaDive databases.
$$ \text{Temperature score} =\frac{\text{length(input temperature range} \cap \text{organism temperature range)}}{\text{length(input temperature range)}}$$
$$ \text{pH score} = \frac{\text{length(input pH range} \cap \text{organism pH range)}}{\text{length(input pH range)}}$$
$$ \text{Survivability score} = \text{Temperature score} + \text{pH score}$$
Combined Formula
The Final score used to return an ordered list of organisms is a linear combination of the Resourcefulness and Survivability scores. Currently, the weights for both the scores have been set equal to ‘1’.
$$ \text{Final score} = \text{w1} \cdot \text{Resourcefulness score} + \text{w2} \cdot \text{Survivability score}$$ $$ \text{where w1, w2} = 1 $$
This can further be customized and fine-tuned as per the end user’s requirements, as this provides a trade-off between the constraints supplied by the user.
BacDive and MediaDive
BacDive is a database that provides information about bacterial and archaeal diversity. BacDive’s well documented REST API still posed issues of inconsistent data formats that required extensive error handling to process the data. We get temperature data for an organism from BacDive from which the temperature ranges are calculated.
MediaDive is a culture media database that was often referenced in BacDive entries. We have utilized this database to infer pH physiological data for a given organism from which the pH ranges are calculated.
Graphical Overview

Web Tool
We have created a web tool that provides a simple and accessible way for users to run our software without needing to download or understand complex code. Releasing code is useful for developers, but for non-experts who are not used to setting up code and handling issues, it can be a challenge. By hosting the tool on a Django server with a user-friendly interface, we make it easy for anyone to use without technical setup. This ensures a wider audience can benefit from the tool, focusing on their research rather than technical details, and allows easy access from any device, improving usability and utility.
We can confirm that this website-hosted form of our software is compatible with the following non-exhaustive list of browsers:
-
Google Chrome 129.0.6668.70
-
Safari on macOS (Laptops and Desktops) 18.0
-
Safari on iOS (iPhone, iPad and iPod) 18.0
-
Mozilla Firefox 130.0.1
-
Opera on Desktop 114.0.5282.21
-
Opera on Android 76.2.4027.73374
For other or non-supported browsers, please refer to our CLI version which can be run locally on the user’s device.
Results
We present our software in two forms. One, as a standalone software working on the command line interface (CLI), to enable easy modifications and integrations with new code. Two, as part of a web application created using the Django framework that can be hosted locally to enable user-friendly access to the code, especially useful for an intended target audience without software or programming expertise. We have deployed this code at http://astrolabeiitm.duckdns.org/ to enable quick and easy access via the internet.
Standalone
The standalone software can be found here. This can be used by running main.py after installing the necessary dependencies listed in requirements.txt.
The input interface for the standalone appears like this on the command line:

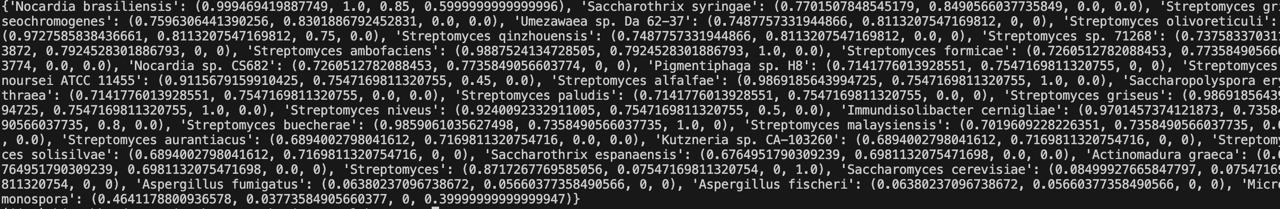
The output is a Python dictionary whose keys are the organism names and values are tuples. Each tuple consists of 4 scores in this order: Final_score, Resourcefulness_score, Temperature_score, pH_score. The organisms are arranged in descending order of their Final_score value.
Online Web Application
We have deployed our software at http://astrolabeiitm.duckdns.org/ to enable quick and easy access via the internet.
Local Host Web Application
To use the Django web application locally, follow these steps: